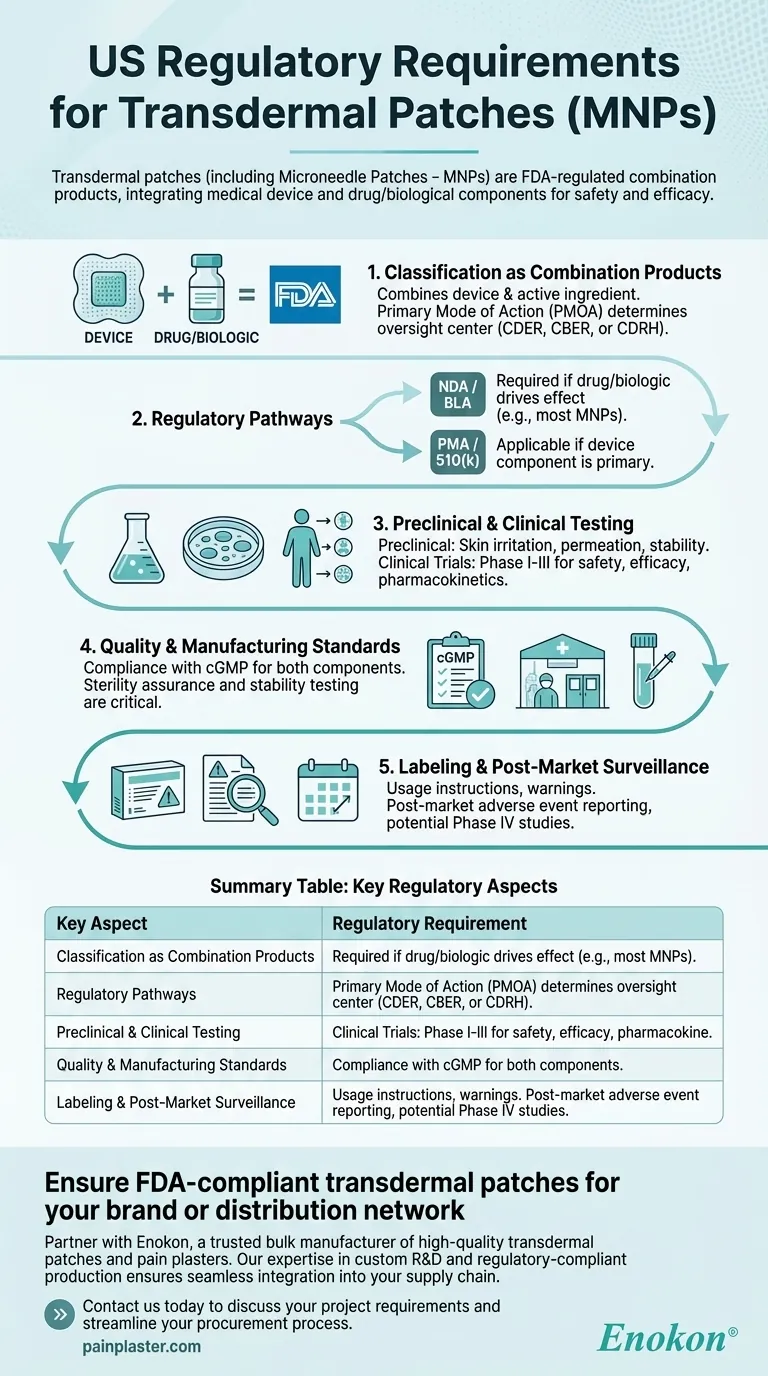
Transdermal patches, including microneedle patches (MNPs), are regulated in the U.S. as combination products by the FDA, requiring rigorous approval processes to ensure safety and efficacy. These products integrate medical device and drug/biological components, necessitating compliance with specific regulatory pathways depending on their primary mode of action. The approval process involves preclinical and clinical testing, quality control, and adherence to labeling and manufacturing standards.
Key Points Explained:
-
Classification as Combination Products
- The FDA categorizes transdermal patches as combination products because they combine a medical device (e.g., patch backing, microneedles) with a drug or biological active ingredient.
- The primary mode of action (PMOA) determines whether the product is regulated by the Center for Drug Evaluation and Research (CDER), the Center for Biologics Evaluation and Research (CBER), or the Center for Devices and Radiological Health (CDRH).
-
Regulatory Pathways
- New Drug Application (NDA) or Biologics License Application (BLA): Required if the drug or biologic component drives the therapeutic effect.
- Premarket Approval (PMA) or 510(k): Applicable if the device component is primary (e.g., microneedles facilitating drug delivery).
- MNPs often follow the NDA/BLA route due to their drug-centric function.
-
Preclinical and Clinical Testing
- Preclinical Studies: Include skin irritation, permeation, and stability testing to assess safety and delivery efficiency.
- Clinical Trials: Phase I-III trials evaluate pharmacokinetics, efficacy, and adverse effects in humans.
-
Quality and Manufacturing Standards
- Compliance with Current Good Manufacturing Practices (cGMP) for both drug and device components.
- Sterility assurance and stability testing are critical for patches with biologics.
-
Labeling and Post-Market Surveillance
- Labels must include usage instructions, warnings, and storage conditions.
- Post-market requirements include adverse event reporting and potential Phase IV studies.
For purchasers, understanding these requirements ensures alignment with FDA-compliant suppliers, reducing risks of non-conforming products. Have you considered how these regulations impact your procurement timelines or vendor selection criteria? The interplay of device and drug oversight quietly shapes the reliability of modern transdermal therapies.
Summary Table:
| Key Aspect | Regulatory Requirement |
|---|---|
| Classification | Regulated as combination products (device + drug/biological) by FDA. |
| Primary Mode of Action | Determines oversight by CDER (drug), CBER (biologic), or CDRH (device). |
| Approval Pathways | NDA/BLA (drug-driven) or PMA/510(k) (device-driven). MNPs typically follow NDA/BLA. |
| Testing Requirements | Preclinical (safety, permeation) and clinical trials (Phases I-III). |
| Manufacturing Standards | cGMP compliance for both drug and device components; sterility assurance for biologics. |
| Post-Market Obligations | Adverse event reporting, Phase IV studies, and labeling compliance. |
Ensure FDA-compliant transdermal patches for your brand or distribution network
Partner with Enokon, a trusted bulk manufacturer of high-quality transdermal patches and pain plasters. Our expertise in custom R&D and regulatory-compliant production ensures seamless integration into your supply chain.
Contact us today to discuss your project requirements and streamline your procurement process.
Visual Guide
Related Products
- Menthol Gel Pain Relief Patch
- Icy Hot Menthol Medicine Pain Relief Patch
- Heating Pain Relief Patches for Menstrual Cramps
- Asthma Cough and Pain Relief Patch for Adults and Kids
- Herbal Medicated Anti Diarrhea Patch for Digestive Relief
People Also Ask
- Who should consult a healthcare professional before using pain relief patches? Ensure Your Safety with Medical Advice
- What are the potential side effects of pain relief patches? A Guide to Safe Use & Key Risks
- How do pain relief patches compare to other pain relief methods? Discover Targeted, Long-Lasting Relief
- Can pregnant women use pain relief patches? Your Essential Guide to Safe Pain Management
- How effective are pain relief patches for muscle pain? Target Localized Pain with Transdermal Delivery